Virtual screening for druglike molecules in general – and new agents with a defined pharmacological profile in particular – is an established concept for hit and subsequent lead candidate identification during the early phases of a drug discovery project, sometimes even in situations with much structural uncertainty and potential ambiguity of receptor-ligand interactions (Figure 1). Coined as a term approximately a decade ago, virtual screening today stands for a rather loosely defined collection of computational methods for screening compound prioritization and design. Although many elegant and successful applications of these techniques have been published over the past years, for example those based on automated ligand-receptor docking or machine learning classifiers, it is most noteworthy that there are almost as many methods as are reported practical applications. There is no single best virtual screening technique which has emerged as the tool for hit identification irrespective of the drug target under consideration. While it is evident that the applicability and thus the usefulness of a certain method are target-dependent properties, a peculiar observation is that current prediction software solutions (e.g., for "drug-likeness", pharmacokinetic parameters, target-ligand binding) rarely exceed 80-85% accuracy at best in retrospective tests, and 40-50% as an optimistic estimate in prospective applications. It is therefore only fair to ask for the reasons for this notorious threshold, as it is almost impossible to judge the real value of "just another virtual screening method" being published with a reported accuracy of 85.3% leading to some low affinity hits at a selected target. Simply demanding for "better training data" is insufficient, often unjustified, and cannot be presented as the only solution – despite the well-known deficiencies of primary HTS data and error-prone readouts of some cell-based assays, to name but a few.
So, what are we missing? Which information will be essential for progress in method development? It is the author’s opinion that an answer to this question will have to address at least the following critical points:
Foremost, we cannot expect practical applications of any virtual screening method to produce perfect results simply because no model is perfect (and sometimes expectations are borderline silly: "If virtual screening does not produce novel hits with nanomolar affinity it is useless."). To strive for perfect prediction of biochemical response profiles for a given screening compound is unrealistic and bears the risk of leading to artifactual, over-trained models. While a perfect "local" prediction model for a certain lead series and a defined target certainly is worthwhile building, it simply is unreasonable to expect perfection from any such "global" method (admittedly, the differentiation between local and global models is artificial itself and will not resolve the underlying discrepancy between the expected model quality and a poor representation of molecular objects).
Furthermore, probably all macromolecular drug targets tolerate various structurally diverse ligand chemotypes, which as yet has not been sufficiently taken into consideration during model development. Future virtual screening methods must be able to account for one-to-many and many-to-many relationships between ligands and targets, which will render them useful for off-target prediction, as well as target and ligand "de-orphanization". Appropriate data from chemo- and pharmacogenomics initiatives should play a key role here and serve as reference.
Finally, as long as entropic contributions to receptor-ligand interactions are grossly neglected, sustained success by virtual screening will be possible only for a limited set of drug-receptor complexes. While progress is being made for the modeling of water molecules involved in ligand binding, flexible fit phenomena, ligand protonation states in proteinous environments, and quantitative interaction types, reliable entropy estimations remain computationally impracticable for high-throughput virtual screening. New concepts are urgently needed to address this critical issue. This will have to include a fresh view on the existing physically and partly mechanistically motivated molecular force fields. We have to ask ourselves whether the existing approaches are sufficient to accurately describe molecular or atomic interactions between a macromolecular receptor and a ligand and compute free energies of binding. Often this is not the case, and possibly algorithmic and technological advances will enable quantum chemical treatment of large systems and molecular complexes as a replacement of current force field approximations in the future.
Another frequently discussed but still insufficiently addressed problem in virtual screening is the stereo-selectivity of targets. To date, this pivotal property of macromolecular receptors has not been satisfactorily addressed in the absence of a reliable target structure model. Many of the established ligand-based virtual screening techniques ignore stereochemistry and might therefore present themselves unsuitable as generic approaches to hit-to-lead optimization. But even current three-dimensional ligand docking and pharmacophore-based methods largely fail to appropriately capture and describe stereo-centers and their preferred conformational ensembles.
Possibly, we could learn much more from systematic receptor pocket analysis about how to construct preferred ligands than by mere automated ligand docking (even if perfect scoring functions were available). For example, ensemble shapes and property distributions of "druggable" binding pockets can serve as additional filtering criteria for fast ligand-based virtual screening. In this way, merging of receptor and ligand information might help eliminate false-positives and rescue false-negatives in a virtual screening triage.
While the main problems to overcome on our quest for better virtual screening tools certainly are of chemical nature (e.g. how to represent a molecule, describe pharmacophoric features, or compute energy contributions), there also remains much to be discovered in informatics. Computational chemistry and molecular modeling have a tradition in physics and theoretical chemistry, and only virtual screening applications have explored machine learning methods to a significant extent. After the initial exploration of artificial neural network models in particular, we currently witness an increasing application of so-called kernel methods for method development, e.g. support vector machines. Unquestionably many more already existing algorithms and concepts may be adapted from engineering and computer sciences and taken on for virtual screening purposes – not to construct "just another virtual screening tool" but to provide an appropriate mathematical framework that is actually able to capture advanced chemical knowledge. Smart combinations of innovative machine learning approaches and advanced, innovative molecular modeling concepts might be suited to help overcome some of the limitations of current virtual screening approaches.
By Gisbert Schneider, Goethe-University
Address of the author:
Dr. Gisbert Schneider
Professor of Chem- & Bioinformatics
Beilstein Endowed Chair for Cheminformatics
Goethe-University
Siesmayerstr. 70
D-60323 Frankfurt am Main, Germany
Email: gisbert.schneider@modlab.de
URL: www.modlab.de
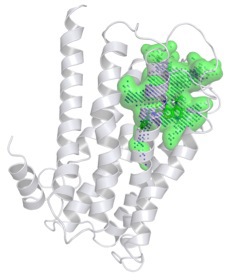
Figure 1. Model building and virtual screening can help generate ligand binding hypotheses and suggest practical validation experiments. The example presents a predicted ligand binding region (green) in a homology model of human histamine H4 receptor, which was obtained by molecular dynamics simulation (Tanrikulu et al., ChemMedChem 2009, 4(5):820-827.).




