|
|

Danijel Kikelj |
LAB PRESENTATION
The Medicinal Chemistry Programme Group at the University of Ljubljana, Faculty of Pharmacy, Slovenia
Mission of the programme group Medicinal Chemistry at the University of Ljubljana - Faculty of Pharmacy (www.ffa.uni-lj.si/en.html), leaded by Danijel Kikelj and comprising several research labs, is a high-quality research in medicinal chemistry directed towards discovery of novel biologically active compounds for human health. In order to fulfil this mission, the programme team is engaged in design, synthesis and biological evaluation of bioactive compounds, in development of new approaches to drug design and synthesis, and in development of novel molecular tools for studying the action of bioactive compounds on molecular level.
The programme group, financed by grants from the Slovenian Research Agency (www.arrs.gov.si/sl/), is becoming increasingly engaged in EU projects. The Framework 6 project INTAFAR (Inhibition of New Targets for Fighting Antibiotic Resistance), aiming at better understanding of the physiology and biochemistry of bacterial cell morphogenesis and peptidoglycan biosynthesis was successfully finished in 2010 (www.eur-intafar.eu). In 2010 the programme group started with a 4-year FP7 EU collaborative project Exploring Marine Resources for Bioactive Compounds: From Discovery to Sustainable Production and Industrial Applications (MAREX) in which marine bioactive compounds are being used as leads for drug design (www.marex.fi). In 2011 the programme team started with the FP7 EU project ORCHID (Open Collaborative Model for Tuberculosis Lead Optimization) which will encompass the parallel progression of the three anti-tubercular compound families through lead optimization and MoA studies for whole cell inhibitors and the optimization of an InhA inhibitor for later preclinical development (http://cordis.europa.eu/home_en.html).
The interdisciplinary conceived research programme Medicinal Chemistry which is based on a uniform concept which comprises (i) understanding the biomolecular basis of disease and (ii) knowing the 3D structure of biological macromolecules (enzymes, receptors) involved in particular disease, is focusing on (a) rational design and discovery of drug molecules exerting their action on validated biological targets, (b) their synthesis and (c) biological evaluation aiming at discovery of innovative medicines with antimicrobial and antiviral, antithrombotic and antitumour activity. A new paradigm of designed multiple ligands targeting two or more biological macromolecules is being applied as an innovative approach to the design of antithrombotic, antibacterial and antitumour drugs. A constituent part of the programme is genomic research which is concentrating on studying influence of compounds, designed and prepared following the outlined concepts, on expression and interactions of proteins in the cell. The aims of this strategy are new innovative bioactive compounds with a potential to be developed to drugs and understanding of their complex action on protein network in the cell.
With the aim of achieving therapeutical benefit in bacterial, viral, thrombotic and cancer diseases, our research is concentrated on (i) bacterial enzymes involved in intracellular steps of peptidoglycan biosynthesis and enzymes which are anti-tubercular targets (ii) enzymes involved in the process of blood coagulation and other serine proteases involved in apoptosis (iii) enzymes involved in metabolism of steroid hormones, (iv) fibrinogen and vitronectin receptors and (v) receptor DC-SIGN of dendritic cells. The dogma of achieving the possibly highest selectivity on particular target still prevailing drug design is being critically confronted with emerging concept of designed multiple ligands which achieve better therapeutic effect by simultaneous modulation of several target macromolecules. Combination of enzyme inhibitory action and receptor modulation activity in a single molecule with the aim of achieving therapeutical benefit represents an outstanding challenge for the research programme. The first successful steps towards integration of receptor antagonist and enzyme inhibitor in the same molecule, still being in the initial stage worldwide, has been already done by our group in antithrombotic compounds and we are striving to develop this concept into an established strategy also in other therapeutic fields. For rational design of drugs targeting particular biological macromolecules we are using molecular modeling tools and developing a concept of mimicking biologically active peptides, sugars and lipids with peptidomimetics, glycomimetics and lipidomimetics, which still remains a leading strategy in drug design starting from natural lead compounds.
Based on the presented uniform concept, the programme comprises the following main research topics:
Inhibitors of peptidoglycan biosynthesis
The biosynthesis of peptidoglycan is a complex process which is carried out in three distinct cell phases (cytoplasmic, membrane and periplasmic) and which involves over 20 enzymes. Due to the essential character of peptidoglycan, action of specific inhibitors of its biosynthesis during bacterial growth rapidly leads to the destructuration of the envelope and to cell lysis. The numerous enzymes involved in this biosynthetic pathway thus constitute potential targets for the search for new antibacterial drugs. Several inhibitors of biosynthesis of the peptide part of peptidoglycan, catalyzed by Mur ligases (MurC, MurD, MurE and MurF) as well as D-alanine-D-alanine ligase (Ddl), have recently been designed and synthesized by our programme group (Figure 1). We try to apply the multiple inhibition concept to ATP-dependant Mur enzymes with a hope that a succesful inhibition of ATP binding to the ATP-binding site could, if a problem of selectivity against other ATP-dependant enzymes such as human kinases would be successfully solved, lead to effective universal chemoterapeutics.
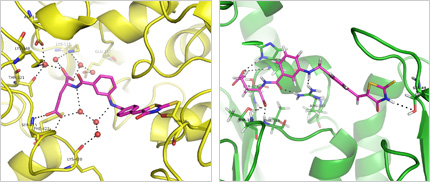
Figure 1. X-ray binding mode of dual inhibitor (IC50 S. aureus MurD = 8,2 µM; IC50 S. aureus MurE = 17,5 µM) possessing antibacterial activity (MIC S. aureus and MRSA = 8 µg/mL) (in magenta) in the active site of MurD (PDB ID code: 2Y1O) and GOLD-calculated conformation of the same inhibitor in MurE model active site (in magenta). Hydrogen bonds between dual inhibitor and MurD and MurE active site residues or crystal water molecules (in red) are presented as dashed lines (Tomašić et al., unpublished results).
Selected recent publications:
- TOMAŠIĆ T, ZIDAR N, ŠINK R, KOVAČ A, BLANOT D, CONTRERAS-MARTEL C, DESSEN A, MÜLLER-PREMRU M, ZEGA A, GOBEC S, KIKELJ D, PETERLIN-MAŠIČ L. Structure-based design of a new series of D-Glutamic acid-based inhibitors of bacterial UDP-N-acetylmuramoyl-L-alanine:D-glutamate ligase (MurD). J Med Chem 2011, 54, 4600-4610.
- TURK S, VERLAINE O, GERARDS T, ŽIVEC M, HUMLJAN J, SOSIČ I, AMOROSO A, ZERVOSEN A, LUXEN A, JORIS B, GOBEC S. New noncovalent inhibitors of penicillin-binding proteins from penicillin-resistant bacteria. PLoS one 2011, 6, e19418.
- TOMAŠIĆ T, KOVAČ A, KLEBE G, BLANOT D, GOBEC S, KIKELJ D, PETERLIN-MAŠIČ L. Virtual screening for potential inhibitors of bacterial MurC and MurD ligases. J Mol Model 2011, doi: 10.1007/s00894-011-1139-8.
- SOSIČ I, BARRETEAU H, SIMČIČ M, ŠINK R, CESAR J, ZEGA A, GOLIČ GRDADOLNIK S, CONTRERAS MARTEL C, DESSEN A, AMOROSO A, JORIS B, BLANOT D, GOBEC S. Second-generation sulfonamide inhibitors of d-glutamic acid-adding enzyme: activity optimisation with conformationally rigid analogues of d-glutamic acid. Eur J Med Chem 2011, 46, 2880-2894.
- ZIDAR N, TOMAŠIĆ T, ŠINK R, ŠKEDELJ V, KOVAČ A, TURK S, PATIN D, BLANOT D, CONTRERAS MARTEL C, DESSEN, A, MÜLLER-PREMRU M, ZEGA A, GOBEC S, PETERLIN-MAŠIČ L, KIKELJ D. Discovery of novel 5-benzylidenerhodanine and 5-benzylidenethiazolidine-2,4-dione inhibitors of MurD ligase. J Med Chem 2010, 53, 6584-6594.
- TOMAŠIĆ, T, ZIDAR N, KOVAČ A, TURK S, SIMČIČ M, BLANOT D, MÜLLER-PREMRU M, FILIPIČ M, GOLIČ GRDADOLNIK S, ZEGA A, ANDERLUH M, GOBEC S, KIKELJ D, PETERLIN-MAŠIČ L. 5-Benzylidenethiazolidin-4-ones as multitarget inhibitors of bacterial Mur ligases. ChemMedChem 2010, 5, 286-295.
- HUMLJAN J, KOTNIK M, CONTRERAS-MARTEL C, BLANOT D, URLEB U, DESSEN A, ŠOLMAJER T, GOBEC S. Novel naphthalene-N-sulfonyl-d-glutamic acid derivatives as inhibitors of MurD, a key peptidoglycan biosynthesis enzyme. J Med Chem 2008, 51, 7486-7494.
Antagonists of receptor DC-SIGN as potential antibacterial and antiviral compounds
DC-SIGN (Dendritic Cell-Specific ICAM-3 Grabbing Nonintegrin) is a C-type lectin implicated in the recognition of pathogens and in some of the earliest stages of the infection process. Certain pathogens exploit DC-SIGN in order to bind immune system cells, but circumvent the processes of internalization and degradation. DC-SIGN is thus used as a Trojan horse to invade lymphocytes T CD4+, which is the main HIV invading mechanism. Recently we have designed, synthesized and biologically evaluated several families of DC-SIGN antagonists and developed an assay that measures inhibition of human dendritic cell adhesion on mannan-coated microtiter plates, suitable for screening a large number of compounds, determination of inhibitory constants (IC50) and fast discovery of new DC-SIGN antagonists.
Recent publications:
- OBERMAJER N, SATTIN S, COLOMBO C, BRUNO M, ŠVAJGER U, ANDERLUH M, BERNARDI A. Design, synthesis and activity evaluation of mannose-based DC-SIGN antagonists. Molecular Diversity 2011, 15, 347-360.
- OBERMAJER N, ŠVAJGER U, JERAS M, SATTIN S, BERNARDI A, ANDERLUH M. An assay for functional dendritic cell-specific ICAM-3-grabbing nonintegrin (DC-SIGN) inhibitors of human dendritic cell adhesion. Anal Biochem 2010, 406, 222-229.
- TIMPANO Gabriele, TABARANI G, ANDERLUH M, INVERNIZZI D, VASILE F, POTENZA D, NIETO, Pedro M, ROJO J, FIESCHI F, BERNARDI A. Synthesis of novel DC-SIGN ligands with an [alpha]-fucosylamide anchor. ChemBioChem 2008, 9, 1921-1930.
Inhibitors of steroid hormone metabolism as potential antitumour drugs
Steroid hormones play an important role in the aetiology of hormone-dependent diseases, such as breast, prostate and endometrial cancer, disorders of reproduction, and neuronal diseases. The occupancy of the steroid hormone receptors is regulated mainly by hydroxysteroid dehydrogenases, which convert steroids at positions 3, 11, 17 and 20 of the steroid core, thereby acting as molecular switches. The 17β-hydroxysteroid dehydrogenases (17β-HSDs) modulate the biological potencies of estrogens and androgens by converting inactive 17-keto-steroids into their active 17β-hydroxy-forms (such as estradiol, testosterone and dihydrotestosterone), or vice versa. These enzymes play a key role in hormonal regulation and function in the human and represent emerging therapeutic target for the control of estrogene- and androgene-sensitive cancers. Recently we have discovered that simple coumarines prepared by Suzuki-Miyaura cross coupling reaction significantly inhibit 17β-HSD1 in a recombinant enzyme assay with high selectivity over 17β-HSD2. The best inhibitors in series were 7-phenyl-3-acetyl coumarin derivatives with the most potent compound having IC50 value of 268 nM and good selectivity over 17β-HSD2 receptors.
Recent publications:
- STARČEVIĆ Š, BROŽIČ P, TURK S, CESAR J, LANIŠNIK-RIŽNER T, GOBEC S. Synthesis and biological evaluation of (6- and 7-phenyl) coumarin derivatives as selective nonsteroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1. J Med Chem 2011, 54, 248-261.
- BRUNSKOLE ŠVEGELJ M, TURK S, BRUS B, STOJAN J, LANIŠNIK-RIŽNER T, GOBEC S. Novel inhibitors of trihydroxynaphthalene reductase with antifungal activity identified by ligand-based and structure-based virtual screening. J Chem Inf Mod 2011, doi: 10.1021/ci2001499.
- STARČEVIĆ Š, KOCBEK P, HRIBAR G, LANIŠNIK-RIŽNER T, GOBEC S. Biochemical and biological evaluation of novel potent coumarin inhibitor of 17ß-HSD type 1. Chem Biol Interact 2011, doi: 10.1016/j.cbi.2011.01.002.
- BROŽIČ P, TURK S, LANIŠNIK-RIŽNER T, GOBEC S. Discovery of new inhibitors of aldo-keto reductase 1C1 by structure-based virtual screening. Mol Cell. Endocrinol. 2009, 301, 245-250.
Coagulation enzymes inhibitors and modulators of fibrinogen receptor as novel antithrombotic compounds with dual action
In order to achieve a synergistic antithrombotic effect, simultaneous application of anticoagulant and antiaggregatory drugs (e.g. thrombin inhibitors and fibrinogen receptor antagonists) is frequently applied in clinical practice. Complementary structures of pharmacophores D-Phe-Pro-Arg of thrombin inhibitors and Arg-Gly-Asp of fibrinogen receptor antagonists inspired us to merge both pharmacophores in a low-molecular weight peptidomimetic compounds which inhibit thrombin and act as antagonist of fibrinogen receptor, thus displaying anticoagulant and antiaggregatory activity. Optimization of a class of compounds afforded dual antithrombotic compounds possessing a well balanced activity on both (Figure 2). The best classes of antithrombotic leads with dual function were found to possess antioxidative and radical scavenging activity which synergistically contributes to their antithrombotic potential.
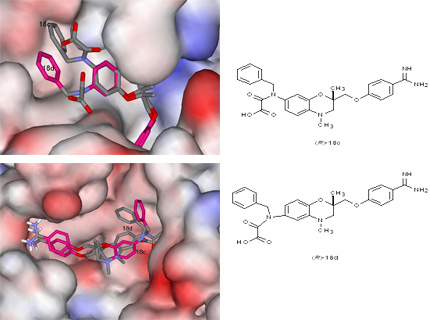
Figure 2. Dual antithrombotic compounds (R)-18c and (R)-18d docked in the active site of thrombin (left) and GPIIb/IIIa binding site (right).
Selected publications:
- ILIĆ M, KONTOGIORGOS C, HADJIPAVLOU-LITINA D, ILAŠ J, KIKELJ D. Thrombin inhibitors with lipid peroxidation and lipoxygenase inhibitory activities. Bioorg Me. Chem Lett 2011, doi: 10.1016/j.bmcl.2011.06.089.
- ILAŠ J, JAKOPIN Ž, BORŠTNAR T, STEGNAR M, KIKELJ D. 3,4-Dihydro-2H-1,4-benzoxazine derivatives combining thrombin inhibitory and glycoprotein IIb/IIIa receptor antagonistic activity as a novel class of antithrombotic compounds with dual function. J Med Chem 2008, 51, 5617-5629,
- ILAŠ J, TOMAŠIĆ T, KIKELJ D. Novel potent and selective thrombin inhibitors based on a central 1,4-benzoxazin-3(4H)-one scaffold. J Med Chem 2008, 51, 2863-2867.
- ŠTEFANIČ P, ANDERLUH M, ILAŠ J, MRAVLJAK J, SOLLNER DOLENC M, STEGNAR M, KIKELJ D. Toward a novel class of antithrombotic compounds with dual function. Discovery of 1,4-benzoxazin-3(4H)-one derivatives possessing thrombin inhibitory and fibrinogen receptor antagonistic activities. J Med Chem 2005, 48, 3110-3113.
Nitroxide-fluorophore double probes
Study of the transmembrane signal transduction mechanism with non-destructive spectroscopic methods [electron paramagnetic resonance (EPR) and fluorescence spectroscopy/microscopy (FS)] is connected with progress in appropriate molecular tools – spin-labels, fluorescence-labels and dual probes. To study extra cellular surfaces, cell interactions with its surrounding and influence of cholesterol oxidized products on membrane domain structure, we try to develop labelled glycomimetics and lipidomimetics which will enable efficient exploitation of all possibilities of non-destructive spectroscopic methods like EPR and FS. A further focus of our research is on design and synthesis of labelled glycomimetics and lipidomimetics with preserved biological activity of non-labelled parent molecule. A special attention is focused on design and synthesis of dual labelled molecular probes that combine paramagnetic nitroxide group and fluorophore. We expect that dual probes will enable simultaneous application of two non-destructive spectroscopic (EPR, FS) techniques and thus make possible more profound understanding of membrane processes.
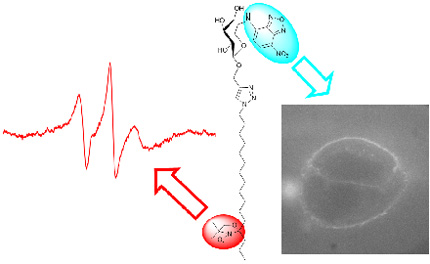
Figure 3. Nitroxide-fluorophore double probes: a potential tool for studying membrane heterogeneity by ESR and fluorescence.
Selected publications:
- PAJK S, GARVAS M, ŠTRANCAR J, PEČAR S. Nitroxide-fluorophore double probes: a potential tool for studying membrane heterogeneity by ESR and fluorescence. Org Biomol Chem 2011, 9, 4150-4159.
- HUMAR M, RAVNIK M, PAJK S, MUŠEVIČ, I. Electrically tunable liquid crystal optical microresonators. Nature Photonics 2009, 3, 595-600.
- PAJK S, PEČAR S. Synthesis of novel amphiphilic spin probes with the paramagnetic doxyl group in the polar region. Tetrahedron 2009, 65, 659-665.
- MRAVLJAK J, PEČAR S. A new glucosamine-containing amphiphilic spin probe. Tetrahedron Lett 2009, 50, 567-569.
Group Members: Danijel Kikelj, Stanislav Gobec, Marija Sollner Dolenc, Aleš Obreza, Lucija Peterlin Mašič, Anamarija Zega, Marko Anderluh, Jožko Cesar, Janez Ilaš, Janez Mravljak, Matej Sova, Rok Frlan, Žiga Jakopin, Matej Živec, Samo Turk, Nace Zidar, Tihomir Tomašić, Roman Šink, Stane Pajk, Irena Mlinarič Raščan, Nataša Karas Kuželički and Damjan Janeš.
For a complete bibliography of the programme group in the last 3 years see http://izumbib.izum.si/bibliografije/P20110721144750-P1-0208.html
Danijel Kikelj and
Lucija Peterlin Mašič
Contact:
Prof. Dr. Danijel Kikelj
University of Ljubljana
Faculty of Pharmacy
Aškerčeva 7
1000 Ljubljana
Slovenia
Phone: +386-1-4769561
Fax: +386-1-4258031
E-mail: danijel.kikelj@ffa.uni-lj.si
|
|

Editor
Gabriele Costantino
Univ. of Parma, IT
Editorial Committee
Erden Banoglu
Gazi Univ., TR
Lucija Peterlin Masic
Univ. of Ljubljana, SI
Leonardo Scapozza
Univ. of Geneve, CH
Wolfgang Sippl
Univ. Halle-Wittenberg, DE
Sarah Skerratt
Pfizer, Sandwich, UK

Executive Committee
Gerhard F. Ecker President
Roberto Pellicciari Past Pres.
Koen Augustyns Secretary
Rasmus P. Clausen Treasurer
Javier Fernandez Member
Mark Bunnage Member
Peter Matuys Member

|

