|
|

Chris de Graaf |
YOUNG RESEARCHER
Chris de Graaf
Most meritorious runner up of the EFMC Prize for a Young Researcher in Academia 2012 and 2013
In silico veritas: Medicinal chemistry through the 3D looking glass
My interest in investigating the structure and function of pharmacologically relevant protein systems originates from my study Chemistry at the University of Amsterdam (1997-2002). The drive to perform scientific research and study the molecular determinants of protein-ligand interactions aligned with an interesting PhD position in the field of computational medicinal chemistry and toxicology at VU University Amsterdam. My PhD project (under the supervision of Prof. Nico Vermeulen) was part of a multidisciplinary research program that combined computational chemistry, organic chemistry, biochemistry, spectroscopy, and molecular toxicology. From 2002 to 2006 I developed complementary computational approaches to study ligand binding to cytochrome P450 enzymes, and experienced the challenges, strengths, and joys of working in an interdisciplinary research team (in particular with my scientific P450 partner in crime Dr. Peter Keizers). I furthermore learned about the power of international scientific collaboration during gratifying research internships in the groups of Prof. Gerd Folkers at ETH-Zurich (to work on molecule docking methods) and Prof. Rebecca Wade at EML Heidelberg/HITS (on advanced molecular dynamics simulation techniques).
Following my PhD, I was eager to further spread my scientific wings and to explore research in a pharmaceutical industry setting. A post-doc position in the Structural Chemogenomics group of Dr. Didier Rognan at the University of Strasbourg in collaboration with AstraZeneca Pharmaceuticals offered me the opportunity to do just that. I was working closely together with scientists from different AstraZeneca R&D sites in the US, UK, and Sweden (including Dr. Ola Engkvist, Dr. Igor Shamovsky, and Dr. Fabrizio Giordanetto), and the computational models and methods I developed were successfully applied in drug development projects to discover and optimize new GPCR ligands. In between the project milestones and work visits to AstraZeneca R&D sites, the stimulating environment in Didiers group offered me the freedom to perform more fundamental scientific studies on the development of new G Protein-coupled receptor modeling and virtual screening techniques. For example, we performed one of the first studies to predict functional selectivity of GPCR ligands using an agonist customized model based on the first antagonists bound crystal structure of a human GPCR (the beta-2 adrenergic receptor)1 and conducted the first successful structure-based virtual screening study to identify small allosteric modulators of class B GPCRs.2 During my post-doc abroad, I did not only appreciate best of both worlds professionally (academia and industry), but also enjoyed the French joie de vivre with a touch of Alsacian Gründlichkeit: From vélo-tout-terrain trips up the flanks of the Vosges, amidst vineyards and deserted pine forests, towards a rewarding Gewürztraminer-Münster-apéro.
Medicinal Chemistry in Amsterdam: Interdisciplinary scientific teamwork
At the end of 2008, Dr. Iwan de Esch and Prof. Rob Leurs convinced me to join the Division Medicinal Chemistry at VU University Amsterdam to lead the computational medicinal chemistry team as an assistant professor. The Division Medicinal Chemistry (headed by Prof. Rob Leurs) combines computer-aided drug design and chemical synthesis (Dr. Iwan de Esch, Dr. Maikel Wijtmans, and myself) with molecular pharmacology and biochemistry (Prof. Martine Smit, Dr. Henry Vischer, Dr. Marco Siderius) to understand the molecular details of ligand-receptor interactions, to elucidate receptor signaling networks, and to use this knowledge for the computational design and synthesis of new bioactive molecules. We work on a diverse set of pharmaceutically relevant protein targets, including G protein-coupled receptors (in particular histamine and chemokine receptors), as well as ligand-gated ion channels, kinases, and phosphodiesterases. All are important targets in the development of drugs against for example inflammation, cancer, neuronal disorders, and neglected tropical diseases.
In this interdisciplinary research environment my team and I have developed chemoinformatics tools and modeling protocols that allow explicit incorporation of experimental data and, vice versa, are used to steer medicinal chemistry programs. Current methods that we use in our studies include ligand-, protein-, and protein-ligand interaction fingerprint based virtual screening methods (e.g. EDprints3, FLAP4 (with Prof. Gabriele Cruciani, University of Perugia), IFP5 (with Dr. Didier Rognan) Snooker6 (with Prof. Jacob de Vlieg, Netherlands e-Science Center)). In the past years we have furthermore constructed several structural chemogenomics databases to navigate protein-ligand interaction space of kinases (KLIFS)7, PDEs (PDEStrIAn), and aminergic GPCRs8. My group has built a track record in protein modeling and virtual screening, exemplified by winning an international competition (GPCR DOCK 2010)9 to predict the three-dimensional coordinates of a ligand bound protein crystal structure (of the CXCR4 chemokine receptor), and by obtaining the highest structure-based virtual screening hit rate to discover novel biologically active molecules (for the histamine H1 receptor, with Prof. So Iwata (Imperial College)).5
Computational chemists can (and, in my opinion, should) play an important role as bridge builders between experimental research disciplines. For most of our medicinal chemists, computer-aided drug design techniques are essential tools to optimize molecules. In our medicinal chemistry group there is no rational ligand design without a pharmacophore or protein-ligand interaction model. Molecular modeling simulations offer insights into protein-ligand interactions at an atomic level. These three-dimensional in silico models guide our molecular pharmacologists to probe protein-ligand binding pockets even further with, for example, site-directed mutagenesis studies.10-11 This iterative multidisciplinary teamwork within the Medicinal Chemistry Divison is highly inspiring.
In 2009 I obtained a prestigious Veni Grant from the Netherlands Organization for Scientific Research (NWO) to set up a research line in the computational prediction of protein-ligand interactions. My team has developed and applied new in silico methods to discover ligands for various proteins and to predict protein-ligand selectivity in order to design better drugs with fewer side effects.4-5,12-14 Together with my colleagues in the Medicinal Chemistry Division at VU University Amsterdam I am currently building a fragment-based chemogenomics platform14 based on in-house experimental fragment screening data, to develop in silico modeling techniques to increase our understanding of the molecular details of protein-ligand selectivity. This platform currently contains not only information and models of intended drug targets (including GPCRs, ligand-gated ion channels, kinases, and phosphodiesterases) but also of undesired drug targets (including cytochrome P450 enzymes and hERG). Our fragment-based chemogenomics approach reduces the molecular complexity that is used to probe different protein binding sites to a level that enables the development of innovative computer models to predict selective protein-ligand interactions. The new computer models will be used for the discovery and design of new ligands with specific protein selectivity profiles (i.e., molecules that are able to modulate one specific protein or multiple therapeutic proteins simultaneously (polyharmacology) but do not interact with off-targets that can cause harmful effects).
These efforts align with the set up of an institutional Chemical Biology platform (under the auspices of our Medicinal Chemistry group) that will link scientific efforts of the Amsterdam Institute for Molecules, Medicines, and Systems (AIMMS) and the VU University Medical Center (VUMC). In the past year I have furthermore been involved in setting up European networks to study G protein-coupled receptors (GLISTEN COST Action CM1207, with Dr. Peter Kolb) and to optimize the binding kinetics of drugs (K4DD Innovative Medicines Initiative (IMI) consortium). These international networks bring together a wide range of complementary methods and expertise that help us and our collaborators to further strengthen and extend the impact of our scientific research. I recently enjoyed participating in such an integrated international scientific research team with the Scripps Research Institute (led by Prof. Ray Stevens), Shanghai Institute for Materia Medica (led by Prof. Ming-Wei Wang), and Novo Nordisk, which reported the first crystal structure of the transmembrane domain of a class B GPCR (the glucagon receptor), and performed extensive mutagenesis studies that enabled the construction of a 3D structural model that predicts how glucagon binds its receptor.15
I consider teaching as a unique and attractive aspect of an academic environment, that is highly complementary to scientific research (and requires at least as much skills and devotion). The many motivated bachelor and master students in our study programs make me eager to learn new things every day. Moreover, I feel privileged to work and have worked with many talented and skilled PhD students and post-docs in the VU computational medicinal chemistry team. I would like to thank all current and former (guest) team players (in particular Albert Kooistra, Luc Roumen, Enade Istyastono, Sabine Schultes, Francesco Sirci, Marijn Sanders, Oscar van Linden, Chimed Jansen, Sebastiaan Kuhne) for their inspiring achievements. These talented young scientists as well as my other (ex-)colleagues and collaborators definitely deserve their fair part of the scientific credits I received as most meritorious runner up for the EFMC Prize for a Young Medicinal Chemist in 2012 and 2013.
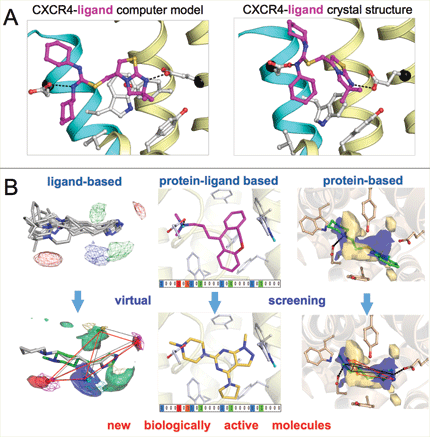
Figure: Examples of the in silico prediction of protein-ligand interactions by the computational medicinal chemistry team of Chris de Graaf: A) In the worldwide GPCR DOCK 2010 competition9 the VU-MedChem team of de Graaf constructed a computational three-dimensional model that correctly predicted the highest number of protein-ligand contacts of all 103 submissions from 25 research groups (prior to release of the experimentally determined CXCR4-ligand co-crystal structure). B) De Graaf and colleagues use ligand-based and protein-based molecular fingerprints for virtual screening of large chemical libraries to discover novel small molecule modulators of pharmaceutically relevant proteins.2,4-5,11-12
References
- de Graaf C, Rognan D. J Med Chem 2008, 51: 4978-4985.
- de Graaf C, Rein C, Giordanetto F, Piwnica D, Rognan D. ChemMedChem 2011, 6: 2159-2169.
- Kooistra AJ, Binsl TW, Van Beek JHGM, de Graaf C, Heringa J. J Chem Info Model 2010, 50: 1772-1780.
- Sirci F, Istyastono EP, Vischer HF, Kooistra AJ, Nijmeijer S, Kuijer M, Wijtmans M, Mannhold R, Leurs R, de Esch IJP, de Graaf C. J Chem Info Model 2012 52: 3308-3324.
- de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, Shimamura T, Iwata S, Stevens RC, de Esch IJP, Leurs R. J Med Chem, 54: 8195-206.
- Sanders M, Verhoeven S, de Graaf C, Roumen L, de Vlieg J, Klomp J. J Chem Info Model 2011, 51: 2277-2292.
- Van Linden OPJ, Kooistra AJ, Leurs R, de Esch IJP, de Graaf C. J Med Chem, doi: 10.1021/jm400378w
- Kooistra AJ*, Kuhne S*, de Esch IJP, R. Leurs, de Graaf C. Br J Pharmacol 2013, 170: 101-126.
- Kufareva I, Rueda M, Katritch V; GPCR Dock 2010 participants {incl. VU-MedChem team of de Graaf et al.}, Stevens RC, Abagyan R. Structure 2011, 19: 1108-1126.
- Istyastono, E.P., Nijmeijer, S., Lim, H.D., van de Stolpe, A., Roumen, L., Kooistra, A.J., Vischer, H.; de Esch, I.J.P., Leurs, R., de Graaf, C. J Med Chem 2011, 54: 8136-8147.
- Schultes S, Nijmeijer S, Engelhardt H, Kooistra AJ, Vischer HF, de Esch IJP, Haaksma EJ, Leurs R, de Graaf C. MedChemComm 2013, 4: 193-204.
- Richter L, de Graaf C, Sieghart W, Varagic, Z, Mörzinger, M, de Esch IJP, Ecker GF, Ernst M. Nature Chem Biol 2012, 8: 455-464.
- Jansen C, Wang H, Kooistra AJ, de Graaf C, Orrling K, Tenor H, Seebeck T, de Esch IJP, Ke H, Leurs R. J Med Chem 56: 2087-2096.
- de Graaf C, Vischer HF, de Kloe GE; Kooistra AJ, Nijmeijer S, Kuijer M, Verheij MHP, England P, van Muijlwijk-Koezen, JE, Leurs R, de Esch IJP. Drug Discovery Today 2013, 18: 323-330.
- Siu FY, He M, de Graaf C, Yang D, Zhang Z, Zhou C, Han GW, Xu Q, Wacker D, Joseph JS, Liu W, Lau JF, Cherezov V, Katritch V, Wang M-W, Stevens RC. Nature 2013, 499: 444-449.
|
|

Editor
Gabriele Costantino
Univ. of Parma, IT
Editorial Committee
Erden Banoglu
Gazi Univ., TR
Lucija Peterlin Masic
Univ. of Ljubljana, SLO
Leonardo Scapozza
Univ. of Geneve, CH
Wolfgang Sippl
Univ. Halle-Wittenberg, DE
Sarah Skerratt
Pfizer, Sandwich, UK

Executive Committee
Uli Stilz President
Koen Augustyns President Elect
Hein Coolen Treasurer
Gabriele Costantino Member
Jordi Gracia Member
Phil Jones Member

|

