|
|

Graeme Robertson |
Perspective
Chemistry at the Biology/Disease Interface
Graeme Robertson
Department of Chemistry and Drug Technologies
University of Perugia
Via del Liceo 1
06123 Perugia
Scientific Advances
The science of medicinal chemistry is advancing rapidly and the availability of data at the chemistry biology interface allows medicinal chemist to design new compounds with ever more data. The overall process of drug discovery however requires significant change to become a more sustainable endeavour. There are many aspects amongst the evolving role of medicinal chemistry to consider from potentially encompassing different molecule types to moving closer to biology and better integrating chemistry and biological data in order to chemically navigate efficiently in biological space to better design effective molecules.
Drug discovery is however still and will most likely remain a largely discipline based, i.e. bio-centric or chemo-centric. An arena in which, chemists and biologists “see” the challenges and problems of understanding biology, its connection to cellular function, and how to modulate these effects from very different perspectives. Key to the medicinal chemist’s role is the need to understand the chemical basis for changes in biological systems and disease pathobiology to thus design molecules with the properties needed to probe target modulation in a disease context. To best achieve this, a more open or less discipline-bounded, approach is required potentially also broadening the chemical space considered of interest to medicinal chemistry. 1
The progress in the field of GPCRs, for example, illustrates very well the recent scientific progress and the better availability of data for medicinal chemists. GPCRs are the largest protein family targeted by small molecules and remain a mainstay of drug discovery. Many of the ligands developed such as, the antipsychotic agents dopamine D3 receptor antagonists evolved from an initial knowledge of the binding (and kinetics) of the “original” natural ligand and the use of phenotypic assays. Most chemists who have worked in the past on dopamine receptors would marvel at the recent availability of a co-crystal structure of the D3 receptor with eticlopride and the possibilities for structure-based design. 2
How general this will become remains to be see but GPCRs are benefiting from a range of scientific advances that is promoting the investigation of their effects not only at a target class level but also to now drive this from a structural perspective. Site-directed mutagenesis and modelling of GPCRs has long played a role in ligand design, but the increasing availability of structural data is allowing better analysis of ligand-protein interactions and the investigation of state-dependant protein conformations (agonist, antagonist, etc.). A range of different receptor templates (X-ray structures) are now available for GPCR homology modelling in addition the thermal stabilisation of GPCRs allows the isolation of specific receptor conformations to elucidate physiologically relevant receptor conformations. For example, both adenosine A2A and muscarinic M1 receptors have been stabilised receptors (StaRs) in the inverse agonist conformations and used to profile association and dissociation rates of antagonists. 3
Recent results on high-resolution NMR study of rhodopsin II suggest that aminergic GPCRs could also be accessible using solution NMR techniques, potentially allowing a more dynamic analysis of receptor conformational variation and the impact of ligand-receptor interactions. 4
Chemistry at the Biology/Disease Interface
Notwithstanding these great scientific advances and the contextual richness of data available to medicinal chemists at a target level, the overall drug discovery process in which they work needs serious revision.
Chemistry (medicinal chemists) needs to play a major role in the process of target validation (target modulation) and in the development of more predictive animal models (data sharing). Both these areas are compounded by the relatively poor understanding of the underlying pathophysiology and disease mechanisms, particularly in humans coupled with a need to navigate biological space from a chemistry (modulator) perspective or viewpoint. Bringing the data together necessary to find new targets across disciplines, i.e. Medicinal Chemistry to Chemical Biology and back again, allows an emphasis on target modulation from project inception to clinical studies in a more complete biological/disease context. Progressing compounds by compound effect relationships, phenotypic profiling, imparts a need for chemists to develop new ways of visualising and using more complex multi-dimensional data to design novel therapeutic agents.
In order to achieve this high quality proof-of-concept compounds (probes) are required that facilitate target validation. 5 Target validation requires modulation of protein signalling, preferably with temporal control, in a disease context. This may well require medicinal chemists to apply their skill sets to non-small molecules that best allow investigation of target modulation in the appropriate setting (biological system) and to help drive novel drug discovery away from single compound – single target analyses and perhaps also finally into more novel target space. 6
Taking neurodegeneration as an example of target modulation in a disease framework illustrates many of the challenges in being able to monitor the effects of compounds in complex systems and use data to design new ligands. Neurodegeneration features a complex interaction of challenges to the proteostasis network in the brain leading to; protein misfolding, inflammation, mitochondrial dysfunction, and oxidative damage. This proteostasis landscape deteriorates with time and many neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s disease are characterized by the appearance of protein deposits, aggregates, plaques that constitute key elements in the disease pathobiology. 7 Confronting unbalance in the proteostasis network with small molecules and interpreting changes in this network at a molecular level is a tough challenge for medicinal chemists. It can’t be expected that a single compound/target approach could improve all aspects of proteotoxicity, and monitoring of the influence of compound treatment across cellular functions is needed rather than focus on say only metabolism or neuroinflammation. Ligand-target effects need to consider ligand-network perturbations, rather than ligand-target or pathway effects.
We’re accustomed to see links in biology at the signalling or disease level, but compound trends are usually displayed within a target or target-class environment rather than a Compound Effect Relationship. 8 For example, typical kinase inhibitors are ATP-binding site ligands and kinases are designated as a “family” based on their ligands greatly facilitating a systematic exploration of kinase-space. 9 Classification of kinases via the ATP-binding site had been one of the best examples of systems-based research 10 but consideration of kinase effector domains would provide a very different route to analysing and exploiting this family. The protein-protein interactions of kinases and their effector proteins remains perhaps a future challenge of chemical biology and the use of both chemistry to advance a molecular understanding of biology. 11
The perturbation of biological systems to gain a more holistic understanding of ligand-target interactions with complex biological systems via linking chemogenomics with systems chemical biology could be one of the answers. 12 The modelling of signalling specificity or redundancy is difficult and transferring the information to drug design data can be even more problematic. Here perhaps a good example is the recent demonstration of the chemical dissection of mitochondrial oxidative phosphorylation (OXPHOS) and its application to screening/profiling compound collections across signalling processes to better understand (mitochondrial) biology and toxicity. Screening across 4 cell-based assays of OXPHOS physiology with multiplexed measurements of nuclear and mitochondrial DNA gene expression revealed several complexities of mitochondrial modulation, including that (i) protein synthesis inhibitors can decouple coordination of nuDNA and mtDNA transcription and that (ii) a subset of HMG-CoA reductase inhibitors, combined with propranolol, can cause mitochondrial toxicity, yielding potential clues about the aetiology of statin myopathy. 13 A recent example of a network approach to target identification and new applications for known compounds or mechanisms has been reported via the integration of phenotypic and chemical indexes in pharmacological space and protein-protein interactions in genomic space. 14 The use by medicinal chemists of such compound (drug) biological profiles or fingerprints calls for a more away from single target SAR but also calls for innovative ways by which to portray the information and visualise the factors influencing these “compound effect relationships”. In other words how best to translate knowledge into innovation-based drug discovery.
Animal models play a central role in translating basic biology into a disease understanding; an excellent recent example was the use xenograft models to offer an explanation of the development of resistance to the glioblastoma chemotherapy, Temozolomide (TMZ). Data from this model and in vitro experiments demonstrated that long-term treatment of astroglioma with TMZ induces increased expression of GLUT/SLC2A transporters, mainly GLUT-3, and the pro-proliferative AKR1C phase 1 drug-metabolizing enzymes that lead to increased resistance. Targeting of GLUT-3 in GBM and/or AKR1C proteins could thus delay the acquisition of TMZ resistance. 15 Experimental animal models should however connect screening environments and data, to readouts used in the clinical setting and critically evolve using results from downstream activities so that compound design and modification can become more predictable and based on knowledge better connect to the human disease context. The depth of knowledge (use) of a given model can however be restricted simply by the limited number of compounds and compound types screened in a given organisation. Much could be gained via the collation of animal model data from larger compound (data) sets across organisations to give statistically more relevant data and hopefully models that are more predictive of clinical effects. Such a move would require a more open approach to data sharing although there are some initiatives underway many within the Innovative Medicines Initiative (IMI) to address at least some of the issues such as creating knowledge infrastructures that enable integration of chemical and biological data (Open Pharmacological Space) and drug safety databases (eTOX). 16 Sharing drug safety and toxicology data or being able to search safety/toxicology papers by similarity searching even would provide great reinvestment and help drive the flow of data from later stage development and the clinic back to early research. For example the eTOX initiative aims to develop just such a database from legacy toxicology reports and public toxicology data; combining this with in silico strategies and tools aimed at better predicting the toxicological profiles of small molecules in earlier stages of research.
The Need for Change
Obviously a chemical perspective at the whole animal level also needs to be reflected at an efficacy or mechanistic level. Thus in order to realise an approach based on a chemical perspective on disease biology a greater understanding of the basic biology that underpins the disease pathophysiology is necessary. The reductionist nature of the target-based approach and over focussing on subsets of information doesn’t effectively consider the complexity of the chemistry-biology-disease interface which is thus not considered in a holistic manner. As a result a considerable “knowledge gap” has developed between an understanding of the dynamics of target signalling at a cellular, phenotype, and disease levels with many compounds failing because of an insufficient knowledge of the basic biology driving the disease phenotype.
There are many options/alternatives for approaching this knowledge gap and if R&D is to progress to become more sustainable then a more collaborative approach to addressing these gaps and to drive innovation via academic-industrial, public private partnerships, or other collaborations where more of the basic underlying biology is utilized for compound effect relationship and their use in compound design are needed. How to translate this knowledge (basic research) into better approaches to drug discovery is therefore a key current consideration, critically a substantial improvement in validation of new therapeutic targets is required, via ligands that modulate target functions in a temporal and dose-dependent manner. This integration of medicinal chemistry with basic biology may well come about by building a more collaborative environment particularly between academia and industry. Bringing together centres of excellence and promoting basic research could significantly help in gaining sufficient in-depth understanding of disease pathophysiology.
Historically, academia has had three equally important missions: teaching, i.e. transfer of knowledge, research, i.e. discovery of new knowledge, and the translation of academic innovation into industry as a contribution to “knowledge-based economy”. Academia has typically performed the basic research that elucidated the underlying mechanisms of disease and identified promising points of intervention, whereas corporate researchers have focussed on applied research toward the discovery and commercialisation of novel drug treatments. 17 This need for research centres to develop expertise in depth and collaborate most likely goes well beyond current concepts of open innovation and requires a more substantial revamp of the process to and indeed beyond clinical studies. 19
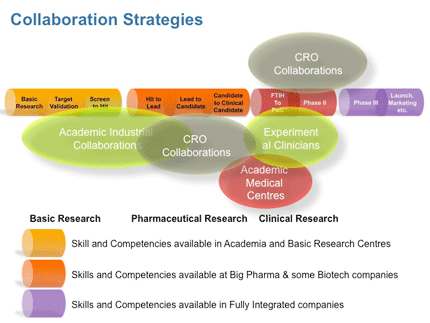
Chemical modulation of signalling networks and cellular events offers opportunities often not accessible with genetic methods, in particular the option for temporal control of cellular events and the development of small-molecule modulators of protein function is at the heart of chemical biology research. It is here that the need to link chemical and biological space and really impact on defining suitable starting points that guide compound design by the recognition of complex structural relationships associated with biological activity. In other words, to use chemical biology to put the “medicinal” back into medicinal chemistry.
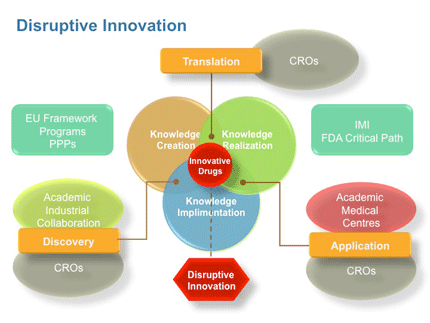
To facilitate this and help drive disruptive innovation it also serves to “simply” bring data together better to permit this chemical navigation of biological and disease space or mapping of the chemistry interfaces. This integration of in-depth basic biology (pharmacology) with cheminformatics and chemogenomic approaches is not however necessarily a “simple” task. 20 Good navigation tools are also required that allow chemists to emerge from rule-based and reductionist approaches to develop a more holistic and knowledge (data) driven approaches. 21 The tools that allow chemical interrogation of biological and disease data should also facilitate the sharing of this across disciplines and organisations. Ultimately, there will be a redefining of the definition of Medicinal Chemistry and a great opportunity for chemists to help drive the next generation of drug discovery. However, chemists need to remain experts at chemistry but diversify to play a wider role in innovative “disruptive” drug discovery, just as centres of excellence need to focus on just that “experts in a particular field collaborating with other experts or centres. 22 This bringing people together to share both contextual and tacit knowledge is often underestimated and more energy needs to be given to the currency or language of collaboration, that is “information sharing”. The direct route to knowledge is not always know or even desired – efficient navigation and interaction is! As stated in the introduction research is still (will remain?) discipline based i.e. bio- or chemo-centric, but the overall drug discovery requires a more open (less bounded) approach. Moreover, generally the more innovative the target the greater the need for collaboration between experts. Such collaboration within networks between centres of excellence (small/large companies and/or academic groups) provides an alternative highly flexible and potentially highly adaptable environment that capitalizes on the collective expertise within the collaborative network. One in which chemists can refine and evolve the role of Medicinal Chemistry.
By Graeme Robertson
_____________________________________________________
References
- Fox D, Sharing our collective wisdom to design better medicines. MedChemWatch 2011, (11), 24-27.
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091-1095.
- Robertson N, Jazayeri A, Errey J, Baig A, Hurrell E, Zhukov A, Langmead CJ, Weir M, Marshall FH. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology. 2010, 60, 36-44.
- Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D. Structure determination of the 7-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat. Struct. Mol. Biol. 2010, 17, 768–774.
- Edwards AM, Bountra C, Kerr DJ, Willson TM. Open access chemical and clinical probes to support drug discovery. Nat. Chem. Biol. 2009, 5, 436-440. Frye, SV, The art of the chemical probe. Nat. Chem. Biol. 2010, 6, 159-161.
- Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH. Too many roads not taken. Nature. 2011, 470. 163-165.
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009, 78, 959-991.
- Paolini GV, Shapland RH, van Hoorn WP, Mason JS, Hopkins AL. Global Mapping of Pharmacological Space. Nat Biotechnol. 2006, 24, 805-815.
- Fedorov O, Müller S, Knapp S. The (un)targeted cancer kinome. Nat Chem Biol. 2010, 6, 166-169.
- Eglen RM, Reisine T. The current status of drug discovery against the human kinome. Assay Drug Dev Technol. 2009, 7, 22-43.
- Augustyn K, Ecker G. Medicinal Chemistry in the 21st Century. MedChemWatch 2010, (10), 5-7.
- Chen B, Dong X, Jiao D, Wang H, Zhu Q, Ding Y, Wild DJ. Chem2Bio2RDF: A semantic framework for linking and data mining chemogenomic and systems chemical biology data. BMC Bioinformatics 2010, 11, 255.
- Wagner BK, Kitami T, Gilbert TJ, Peck D, Ramanathan A, Schreiber SL, Golub TR, Mootha VK. Large-scale chemical dissection of mitochondrial function. Nat Biotechnol. 2008, 26, 343-351.
- Zhao S, Li S. Network-Based Relating Pharmacological and Genomic Spaces for Drug Target Identification. PLoS One 2010, 5, e11764.
- Le Calvé B, Rynkowski M, Le Mercier M, Bruyère C, Lonez C, Gras T, Haibe-Kains B, Bontempi G, Decaestecker C, Ruysschaert JM, Kiss R, Lefranc F. Long-term in vitro treatment of glioblastoma cells with TMZ increases resistance in vivo through up-regulation of GLUT transporter and AKR1C expression. Neoplasia. 2010, 12, 727-739.
- http://www.etoxproject.eu
- Stevens AJ, Jensen JJ, Wyller K, Kilgore PC, Chatterjee S, Rohrbaugh ML. The role of public-sector research in the discovery of drugs and vaccines. N Engl J Med. 2011, 364, 535-541.
- Altshuler JS, Balogh E, Barker AD, Eck SL, Friend SH, Ginsburg GS, Herbst RS, Nass SJ, Streeter CM, Wagner JA. Opening up to precompetitive collaboration. Sci Transl Med. 2010, 2, 52cm26.
- Norman T, Edwards A, Bountra C, Friend S. The Precompetitive Space Time to move the Yardsticks. Sci Transl Med. 2011, 3, 76cm10.
- Campbell SJ, Gaulton A, Marshall J, Bichko D, Martin S, Brouwer C, Harland L. Visualizing the drug target landscape. Drug Discov. Today. 2010, 15, 3-15.
- Oprea TI, May EE, Leitão A, Tropsha A. Computational systems chemical biology. Methods Mol. Biol. 2011, 672, 459-488.
- Hoffmann T, Bishop C. The future of discovery chemistry: quo vadis? Academic to industrial--the maturation of medicinal chemistry to chemical biology. Drug Disc. Today. 2010, 15, 260-264.
|
|

Editor
Gabriele Costantino
Univ. of Parma, IT
Editorial Committee
Erden Banoglu
Gazi Univ., TR
Lucija Peterlin Masic
Univ. of Ljubljana, SI
Leonardo Scapozza
Univ. of Geneve, CH
Wolfgang Sippl
Univ. Halle-Wittenberg, DE
Sarah Skerratt
Pfizer, Sandwich, UK

Executive Committee
Gerhard F. Ecker President
Roberto Pellicciari Past Pres.
Koen Augustyns Secretary
Rasmus P. Clausen Treasurer
Javier Fernandez Member
Mark Bunnage Member
Peter Matuys Member

|

