|
|

Gerhard Ecker |
LAB PRESENTATION
The Pharmacoinformatics Research Group at the University of Vienna
The Pharmacoinformatics Research Group
Originally established as Emerging Field Pharmacoinformatics at the Faculty of Life Sciences at the University of Vienna, the Emerging Field developed towards a full professorship of Pharmacoinformatics. The international team, headed by Prof. Dr. Gerhard Ecker, follows a holistic approach, combining multi-dimensional annotation, structural modeling of biomolecular systems, structure-based drug design, chemometric and in silico chemogenomic methods, statistical modeling and machine learning approaches, to develop predictive computational models. The experimental validation and optimisation of the obtained in silico models by strong links to experimental groups is an integral part of these activities. Currently the targets under investigation include several ABC-transporter (ABCB1, ABCC2, ABCB11, ABCG2), the serotonin and dopamine transporter, the GABA transporters, the hERG potassium channel, the TRPV1 channel, the GABAA receptor, and the insulin receptor. On a methodological basis, the group works on development of new, similarity-based descriptors and the application of unsupervised neural networks for hit-identification.
Ligand-based Studies on ABC-transporters
The main line of research is along the molecular basis of drug-transporter interaction. This started in 1993 with the discovery that the class 1C antiarrhythmic agent propafenone is able to inhibit the multidrug transporter P-glycoprotein (P-gp, ABCB1). P-gp transports a wide variety of functionally and structurally diverse cytostatic agents out of tumor cells, thus preventing accumulation of these drugs within the cells. Inhibition of the transport leads to resensitisation of multidrug resistant cells and thus presents a useful concept for therapy of resistant tumors. Analogous transport systems were also identified in bacteria and fungi. In a strong collaboration with Peter Chiba from the Medical University of Vienna we started a lead optimization program using mainly 2D- and 3D-QSAR studies, which resulted in several tool compounds with potency in the nanomolar range. Besides standard techniques such as Hansch-analysis, Free-Wilson analysis, CoMFA, and GRIND, the group also explores the potential of artificial neural networks, i.e. feedforward backpropagation networks and self organizing maps. The latter have also been successfully applied for scaffold hopping and virtual screening, both for identification of new P-gp inhibitors and insulin receptor activators, as well as for classification of hERG channel blockers.
P-glycoprotein and related transporters are also strongly influencing ADME properties, such as intestinal absorption, blood-brain barrier permeation, and canalicular excretion. Thus, substrates of P-gp and related transporters might face the risk of poor bioavailability, while inhibitors may lead to drug/drug interactions and drug induced cholestasis. The group is thus also actively engaged in developing classification models for prediction of P-gp substrates and inhibitors. In order to mimic real life scenarios, where the number of inactives by far exceeds the number of actives (100:1 to 1000:1), machine learning methods such as random forest and support vector machines, both combined with cost sensitive bagging, are applied for classification of large data sets.
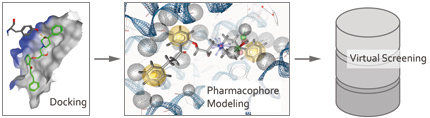
The application of a SAR-guided docking protocol provided a highly reliable binding hypothesis for a class of P-gp inhibitors. Out of this binding mode a structure based pharmacophore model was generated, which in turn was used for virtually screening commercial vendor databases for identifying new P-gp inhibitors.
Experimental Data Guided Ligand Docking
One major focus of the Pharmacoinformatics Research Group is to combine ligand- and structure-based methods. Briefly, the extensive ligand-based in silico studies reveal a clear picture on pharmacophoric sub structures and guide the selection of compounds to be docked into protein homology models of the target under consideration. Subsequent unrestrained docking of these ligands followed by clustering of common scaffolds reveals clusters of binding hypotheses, which are prioritized and validated on basis of the SAR known. As a final prove of concept, top prioritized poses serve as basis for the generation of structure-based pharmacophore models, which are used for screening vendor libraries. Top ranked compounds are ordered and biologically tested. This approach has been successfully applied for identification of new ligands of the benzodiazepine binding site at the GABA-A receptor, inhibitors of P-gp, and inhibitors of the serotonin transporter. The latter two projects are pursued under the framework of the national research cluster SFB35 – “Transmembrane Transporters in Health and Disease” (www.sfb35.at).
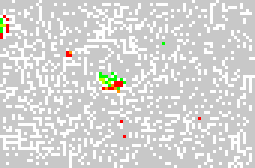
Self-organizing maps as screening tools: Compounds with known activity (green: active, red: inactive) are trained with compounds from a screening database (grey) to find new hits.
Current and Future Challenges
With the increased understanding of physiological and pathophysiological processes driven by systems biology, pharmacoinformatics will move towards pathway driven drug discovery and integrated approaches. First attempts made in our group focus on prediction of cns related side effects of antidepressant agents based on their interaction profiles with a set of 30 transporters and receptors expressed in the cns. Another challenge, the prediction of the outcome of a 28 day rat toxicity study, is pursued under the framework of eTOX, a European project funded by the Innovative Medicines Initative (IMI; www.imi.europa.eu). Finally, by coordinating the Open PHACTS project (www.openphacts.org), the group made a strong move towards semantic integration of public and private data sources relevant for drug discovery. Open PHACTS is also funded by the IMI and aims at developing an Open Pharmacological Space utilising sematic web technology based on rdf triples and nanopublications. This will allow to answer questions such as “give me all compounds which have been annotated with liver toxicity and the interaction profile of these compounds with all transporters expressed in the liver” within a few minutes.
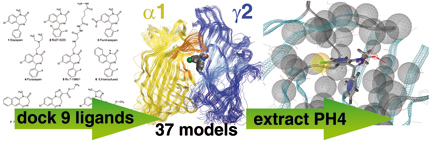
In the search for the binding mode of diazepam at the GABAA-receptor we conducted an exhaustive docking run. We docked a total of nine 5-aryl-1,4-benzodiazepines into 37 homology models of the benzodiazepine binding site of the GABAA receptor, considering flexible side-chains. We retained the 100 best scored poses per docking run. The huge amount of poses was then clustered according to their common 1,4-benzodiazepine scaffold. Following the hypothesis that similar ligands bind in a similar manner, we rejected clusters that could not accommodate essential ligands. The three remaining clusters were heavily evaluated against experimental data resulting in one clear favorite cluster, representing the proposed binding mode. Finally a structure-based pharmacophore was extracted from this final binding mode. This model was then used in a virtual screening study, leading to the identification of a new, experimentally validated, benzodiazepine binding site ligand.

About Gerhard Ecker
Gerhard Ecker is Professor for Pharmacoinformatics and Head of the Pharmacoinformatics Research Group at the Department of Medicinal Chemistry, University of Vienna. He also coordinates the research focus “Computational Life Sciences” of the Faculty of Life Sciences. Gerhard received his doctorate in natural sciences from the University of Vienna and performed his post-doctoral training at the group of J. Seydel in Borstel (Germany). He has published more than 100 articles mainly related to SAR and QSAR studies on P-glycoprotein (P-gp), edited 3 books and gave more than 100 invited lectures. Gerhard is Editor of Molecular Informatics and member of Editorial Advisory Boards and Editorial Boards of several journals. In the years 2009 – 2011 he served as President of the European Federation for Medicinal Chemistry (EFMC), and from 2012 on he chairs the Strategic Advisory Board of EFMC. Finally, he is also strongly engaged in national and international educational activities, such as the national PhD network “Molecular drug targets” (moltag.univie.ac.at) and the EUROPIN PhD Programme in Pharmacoinformatics (www.europin.at), which comprises seven top groups in the field of pharmacoinformatics from 6 European countries.
_____________________________________________________
Contact
Prof. Gerhard F. Ecker
Dept. Medicinal Chemistry, Univ. Vienna
Althanstrasse 14, 1090 wien, Austria
Phone: +43-1-4277-55110
Fax: +43-1-4277-9551
Email: gerhard.f.ecker@univie.ac.at
Lab's website: pharminfo.univie.ac.at
_____________________________________________________
Key publications
- P. Chiba, S. Burghofer, E. Richter, B. Tell, A. Moser, G. Ecker: "Synthesis, Pharmacologic Activity and Structure-Activity-Relationships of a Series of Propafenone-Related Modulators of Multi-Drug-Resistance", J. Med. Chem. 38, 2789-2793 (1995)
- G.F. Ecker, E. Csaszar, S. Kopp, B. Plagens, W. Holzer, W. Ernst, P. Chiba: Identification of ligand-binding regions of P-glycoprotein by activated-pharmacophore photoaffinity labeling and MALDI-TOF mass spectrometry, Mol. Pharmacol. 61, 637-648 (2002)
- Cramer J, Kopp S, Bates SE, Chiba P, Ecker GF. Multispecificity of drug transporters: probing inhibitor selectivity for the human drug efflux transporters ABCB1 and ABCG2. ChemMedChem 2, 1783-1788 (2007)
- Demel MA, Krämer O, Ettmayer P, Haaksma EEJ, Ecker GF. Ensemble Rule-based classification of substrates of the human ABC-transporter ABCB1 using simple physicochemical descriptors. Mol Inform 29, 233-242 (2010)
- Thai K-M, Ecker GF. Classification models for hERG inhibitors by Counter-Propagation Neural Networks. Chem Biol Drug Des 72, 279-289 (2008)
- Thai KM, Windisch A, Stork D, Weinzinger A, Schiesaro A, Guy RH, Timin EN, Hering S, Ecker GF. The hERG Potassium Channel and Drug Trapping: Insight from Docking Studies with Propafenone Derivatives. Chem Med Chem 5, 436-442 (2010)
- Kaiser D, Terfloth L, Kopp S, de Laet R, Chiba P, Ecker GF, Gasteiger J. Self-organizing maps for identification of new inhibitors of P-glycoprotein. J. Med. Chem 50, 1698-1702 (2007)
- C. Tmej, P. Chiba, M. Huber, E. Richter, M. Hitzler, K.-J. Schaper, G. Ecker: "A Combined Hansch/Free-Wilson Approach as Predictive Tool in QSAR Studies on Propafenone-Type Modulators of Multidrug Resistance", Arch. Pharm. Med. Chem. 331, 233-240 (1998)
- Ecker GF, Stockner T, Chiba P. Computational models for prediction of interactions with ABC-transporter. Drug Discov. Today 13, 311-317 (2008)
- Sugano K, Kansy M, Artursson P, Avdeef A, Bendels S, Di L, Ecker GF, Faller B, Fischer H, Gerebtzoff G, Lennernaes H, Senner F. Coexistence of passive and carrier-mediated processes in drug transport. Nature Rev Drug Discov 9, 597-614 (2010)
- Sarker S, Weissensteiner R, Steiner I, Sitte HH, Ecker GF, Freissmuth M, Sucic S. The high-affinity binding site for tricyclic antidepressants resides in the outer vestibule of the serotonin tranporter. Mol Pharmacol 78, 1026-35 (2010)
- Jabeen I, Wetwitayaklung P, Klepsch F, Parveen Z, Chiba P, Ecker GF. Probing the stereoselectivity of P-glycoprotein – synthesis, biological activity and preliminary docking studies of a set of enantiopure benzopyrano[3,4b][1,4]oxazines. Chem Comm, 47, 2586-2588 (2011)
- Klepsch F, Chiba P, Ecker GF. Exhaustive sampling of docking poses reveals binding hypotheses for propafenone type inhibitors of P-glycoprotein. PLoS Comp Biol 7, e1002036 (2011)
|
|

Editor
Gabriele Costantino
Univ. of Parma, IT
Editorial Committee
Erden Banoglu
Gazi Univ., TR
Lucija Peterlin Masic
Univ. of Ljubljana, SLO
Leonardo Scapozza
Univ. of Geneve, CH
Wolfgang Sippl
Univ. Halle-Wittenberg, DE
Sarah Skerratt
Pfizer, Sandwich, UK

Executive Committee
Uli Stilz President
Gerhard F. Ecker Past Pres.
Koen Augustyns Secretary
Rasmus P. Clausen Treasurer
Hein Coolen Member
Gabriele Costantino Member
Phil Jones Member

|

