So why should we use computers in medicinal chemistry? Well, theres so much bioactivity data available which we can (and should) put to good use!
10 Years in the Cheminformatics Field From a Start-Up in Berlin to a Lectureship in Cambridge
There are times in life where you make consciously the right decision - but there are also those situations where a seemingly random choice turns out to be the exactly right one in hindsight. In my case, after studying chemistry at Technical University Berlin and returning from an exchange year at Trinity College Dublin, I had the summer after my year abroad off and having been the decent student I was I decided to look for an internship in a related field. Now, originally I planned to work in South America in summer, but realizing the preparations required for this step I settled for an internship with a cheminformatics start-up close to Berlin instead, in Hennigsdorf, called CallistoGen.
It was the golden time of biotech, back then in summer 2000 (hard to imagine today probably!), and this was the first time that I was in touch with ligand-based approaches to drug design, giving me a scientific direction for about 10 years by now. While admittedly being neither a computer expert nor capturing every detail of the virtual screening algorithms I helped to develop at that time, this stint made me realize my desire to move away from the bench, and to simulations-based work from now on. (An accident in my chemistry lab at home where I nearly lost my eyesight and which kept me in hospital for over a month might have been a contributing factor as well.)
The nice thing about science is that you can move around relatively freely. Hence, for my Masters Thesis I went to Goethe University Frankfurt to work with Gisbert Schneider on a bioinformatics topic; while for my PhD I moved to the UK to extend previous work on virtual screening algorithms at the Unilever Centre for Molecular Informatics in Cambridge / UK with Bobby Glen (which, in fact, is also the place I returned to just a few months back to assume a position as a research group leader). What I am very grateful for is a so-called Presidential Postdoctoral Fellowship with Novartis in Cambridge / MA afterwards it is a fantastic program, which allows the postdoc to do essentially three years of independent research, with all the resources a pharmaceutical company can offer. (Presidential here refers to the head of the research arm of Novartis, not W in case you might wonder.) Given the independent research possible in this position, and under the direction of Jeremy Jenkins, we published more than a dozen papers during my tenure with Novartis, which allowed me to become an Assistant Professor with the Leiden / Amsterdam Centre for Drug Research (LACDR) before assuming my current position as a lecturer with Cambridge University.
Chemogenomics Databases Millions of Bioactivity Datapoints are at our Disposal for Ligand Design Efforts
So why should ligand-based drug design methods be of interest in current days, with more and more crystal structure popping up in databases every day? Well not only that both methods are often complementary, but there is simply such a lot of ligand bioactivity (and more generally, property) data out there which we can (and should) put to good use for future medicinal chemistry activities. To mention a few numbers: The ChEMBL database at the European Bioinformatics Institute (EBI) which was recently released publicly as one of the biggest databases of this type, contains more than 560,000 compound records with more than 2,700,000 experimental activities in its current release, spanning more than 7,300 targets. (Targets in this context might be proteins, but also phenotypic readouts such as as walking behaviour or change in foot-licking latency which are surely some of the more interesting assays to perform). Also other properties, such as lipophilicity or solubility are of interest to the modelling community of course, but I have to say that, personally, I am most interested in the factors that contribute to the activity of chemical matter against protein targets.
This type of data can now be used in multiple ways which are also pursued in my current research group in Cambridge, as well as previously in Leiden, termed chemogenomics or proteochemometrics approaches. These modelling methods take information about the ligand structure, as well as the target protein, into account in order to make bioactivity predictions. So why is this useful? Well, imagine you have a series of compounds, A1, A2, and A3, which you measure against target 1. At the same time, you know the activities of compounds B1, B2 and B3 against target 2 (see Figure 1 for an illustration). In classical models, you would need to generate two separate bioactivity models, one for target 1 and one for target 2, each of which covers only a relatively small area of chemical space each. However, if you knew how to translate bioactivities from target 1 into bioactivities against target 2, you would on the one hand cover more chemical space in your model (namely chemical space represented by all of the ligands above), and on the other hand you would learn to be able to extrapolate between targets 1 and 2. You are now thinking of ligand selectivity (or desired promiscuity) profiles against GPCRs? Or bioactivities against highly resistant mutants of viral enzymes? Then you move into the absolutely right direction - these are the typical areas where proteochemometrics research could be applied, and where we will release prospectively validated primary research results in the very near future.
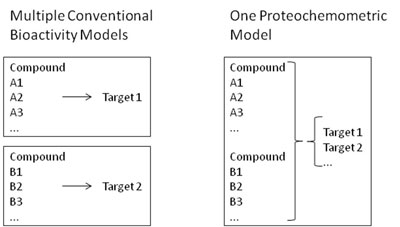
Figure 1. While for conventional bioactivity models one model per activity class needs to be generated, in proteochemometric models only a single model, covering all related protein targets at once, is required. Advantages are larger coverage of chemical space on the ligand side, plus the ability to extrapolate to novel, but related targets (the extent of which depends on the precise data given).
As illustrated in this example, by employing chemogenomics principles namely relating proteins by the similarities of their ligands - we can use ligand bioactivity data in a much wider way then before. This type of modelling, taking into account up to millions of data points, has applications that range from receptor deorphanization, to the prediction of polypharmacology of compounds against G-Protein Coupled Receptors, to the selection of the right HIV Reverse Transcriptase Inhibitor active against a particular mutant of this enzyme.
From Chemical Space, via Protein Targets and Pathways, to Phenotypes (and back again)
One can even go a step further than this, and not only map chemical space onto biological space: The next logical step, and this is also the plan for future research in my group at Cambridge University, is to include information also about phenotypic space in this concept. This would likely involve two steps, firstly pathways information mapped onto the targets, and secondly including also phenotypic information that is known to be related to modulation of those pathways. A database of this type, schematically displayed in Figure 2, could be used in a myriad ways in drug discovery: Be it in the analytical way, by deconvoluting the mechanistic reasons behind adverse drug reactions, or in the way more relevant for drug discovery, by rationally choosing ligand chemical features in order to modulate targets and pathways in a manner to reverse the diseased phenotype. Being realistic, our knowledge of bioactive chemical space and pathway annotations is overall still very limited still, we have millions of chemogenomics data points at our disposal, which we can already use today to make more informed decisions on how to modify compounds to achieve the desired, phenotypic effect

Figure 2. By integrating chemical space with target and pathway annotations, plus a phenotype definition that depends on the particular study, the wealth of data available today can be used in both directions: to rationalize phenotypic observations of compounds (such as adverse reactions), as well as to influence phenotypes the objective of every drug discovery programme.
PhD Students and Postdoctoral Positions Open at the Unilever Centre
The above are only examples of the amount of knowledge and the versatility current bioactivity and pathway databases offer to researchers in the life sciences and I would be very happy to contribute with my experience in chemical data mining and retrieval to experimental ligand design projects in research groups anywhere in the world. Given our recent renewal of funding at the Unilever Centre for Molecular Informatics at the University of Cambridge we are currently heavily recruiting about 10 PhD students and postdoctoral fellows in and around the cheminformatics, chemogenomics and metabolism areas. If you are interested you are cordially invited to visit the Unilever Centre website at http://www-ucc.ch.cam.ac.uk/ or my personal departmental website at http://www.ch.cam.ac.uk/staff/ab.html for further information. Also of course, feel free to contact me directly by e-mail at Andreas.Bender@cantab.net if you would like to discuss options for joint research projects or if you are interested in joining our research groups.






