|
|

Gisbert Schneider |
LAB PRESENTATION
The Computer-Assisted Drug Design Group at ETH Zurich
modlab – The Molecular Design Laboratory
Research activities of the Computer-Assisted Drug Design Group, which was installed in the Department of Chemistry and Applied Biosciences at ETH Zurich in 2010, concentrate on method development for virtual screening, molecular de novo design, and adaptive autonomous systems in drug discovery. In a trans-disciplinary approach the international team amalgamates computer-based pattern recognition and machine learning with compound synthesis and biochemical activity determination. Prior to joining ETH, Prof. Gisbert Schneider was a full professor (Beilstein Endowed Chair for Chem- and Bioinformatics) at Goethe-University in Frankfurt, Germany (2002-2009), where he currently holds a distinguished adjunct professorship, and a researcher with F. Hoffmann-La Roche Pharmaceuticals in Basel, Switzerland (1996-2001).
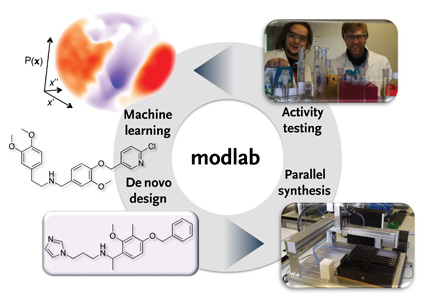
The “modlab team” at ETH conceives, develops and implements new concepts, algorithms and software for rapid identification of bioactive tool compounds and pharmaceutical lead structures. We employ a broad repertoire of computational techniques for automated hypothesis generation, activity prediction and simulation. At the heart of our studies lies the machine-driven de novo design of both individual candidate molecules and small focused compound libraries that exhibit a desired pharmacological activity profile. Research studies also include drug re-purposing, target and off-target prediction, in silico polypharmacology and chemogenomics projects, analysis of protein structure and modulation of protein-protein interaction, as well as the de-orphanization of drugs and their macromolecular receptors. We run own synthesis and testing facilities and a service point for virtual screening (SerViS). In collaborative projects we investigate RNA-protein interactions, and design innovative immunomodulatory agents and anti-inflammatory lead structure candidates. More recently, the scope has been extended to antigen prediction and design, bacterial genome mining for new antimicrobial drug targets, the rational design of host-defense peptides, and the application of computational tools to natural product simulation and the design of natural-product mimicking compounds.
The group has a long-standing track record in molecular de novo design. Since 20 years, we have consequently optimized adaptive algorithms for computer-assisted compound generation by “simulated molecular evolution”. Ten years ago, our ligand-based de novo design software TOPAS (TOPology Assigning System) provided the basis for a fully automated evolutionary molecular design tool. Its youngest descendant, the software DOGS (Design Of Genuine Structures), employs validated chemical reactions and fragment-based building-block assembly for “scaffold-hopping”. De novo designed compounds are analyzed using own software for “fuzzy pharmacophore” matching, chemical landscape analysis, and automated ligand docking. DOGS has recently been applied to generating a novel selective inhibitor of polo-like kinase 1 (Plk1), a target for the development of cancer therapeutics. This pioneering study demonstrates that by coupling of virtual screening, fragment-based chemical synthesis and activity testing, new bioactive agents with a desired target profile can be obtained.
Leads on demand
On the way from models to molecules our algorithms guide an evolutionary design process that constantly adapts to a dynamic fitness landscape (structure-activity function) by integrating new test results that are fed back in iterative synthesis-and-test cycles (active learning concept). Compounds are generated from readily available building blocks by straightforward chemical synthesis in analytical or semi-preparative amounts, and subsequently tested for target binding in vitro. The ultimate goal is to construct an unsupervised molecular design automaton generating “leads on demand”. While the actual realization of this idea might appear futuristic, the overall concept is well motivated and meant to support drug discovery projects by providing innovative technology for the identification of pharmaceutically active agents in a cost- and time-efficient manner. We couple machine-learning with miniaturized synthesis technology and microfluidic lab-on-a-chip devices to prospectively enable broad application of de novo molecular design in medicinal chemistry and explore the full potential of computer-assisted compound optimization.
In tight cooperation with leading groups from academia and pharmaceutical industry, our innovative design concepts are applied and tested for their applicability and usefulness in drug discovery projects. As the molecular design cycle involves multiple scientific disciplines and requires rigorous inter-disciplinary thinking, our team consists of students and researchers with different scientific skills and background. Excellent equipment is available to support computer scientists, bio/cheminformaticians, pharmaceutical chemists, biochemists, and engineers alike. The aim is to provide an ideal research environment for the complete spectrum of computer-assisted drug discovery and break down potential barriers between individual scientific disciplines.
Lead finding in a nutshell A case study
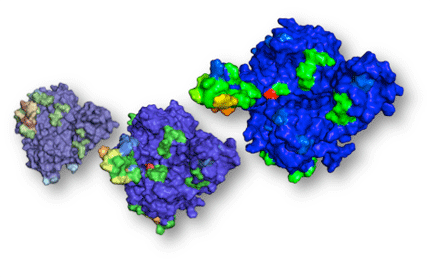
Increasing bacterial resistance against current therapeutic drugs is observed, and novel intervention strategies are urgently sought for. This is also true for the human pathogen Helicobacter pylori (H. pylori), which is responsible for the development of severe gastric inflammation and cancer diseases. In tight cooperation with an expert microbiology team (Prof. S. Wessler, Paris-Lodron University Salzburg, Austria), we analyzed the H. pylori genome by bioinformatics methods and identified the protease HtrA as a novel virulence factor and potential drug target for treatment of bacterial infection. We developed a structure-based virtual screening protocol that starts from the prediction of “hot-spot” surface residues and the automated extraction of a ligand-binding pocket from the protein model. Then, an idealized “virtual ligand” was computed inside this pocket volume, so that pharmacophoric interaction sites between the protein and potential ligand compounds are satisfied. The virtual ligand model finally served as template for rapid virtual screening of compound databases. By using comparative protein modeling and multiple virtual screening techniques, we rapidly identified first-in-class inhibitors of HtrA that efficiently block H. pylori invasion of gastric epithelia This study nicely demonstrates how computational genome mining can lead to novel antibacterial drug target candidates, for which receptor-based virtual screening with a “fuzzy” structure-based pharmacophore model retrieved druglike bioactive agents that combat pathogens.
This outcome and others alike corroborate our trans-disciplinary approach at the interface between theory and laboratory experiment, which proves to be both appropriate and essential for finding inventive solutions to pressing issues in medicinal chemistry.
Gisbert Schneider
Swiss Federal Institute of Technology (ETH)
Department of Chemistry and Applied Biosciences
Institute of Pharmaceutical Sciences
Wolfgang-Pauli-Str. 10
8093 Zürich, Switzerland
Email: gisbert.schneider@pharma.ethz.ch
URL: http://www.modlab.ethz.ch
Selected recent publications:
- Hartenfeller, M. & Schneider, G. (2011) De novo drug design. Methods Mol. Biol. 672, 299-323.
- Schneider, G., Geppert, T., Hartenfeller, M., Reisen, F., Klenner, A., Reutlinger, M., Hähnke, V., Hiss, J. A., Zettl, H., Keppner, S., Spänkuch, S. & Schneider, P. (2011) Reaction-driven de novo design, synthesis and testing of potential type II kinase inhibitors. Future Med. Chem. 3, 415-424.
- Urbanek, D. A., Proschak, E., Tanrikulu, Y., Becker, S., Karas, M. & Schneider, G. (2011) Scaffold-hopping from aminoglycosides to small synthetic inhibitors of bacterial protein biosynthesis using a pseudoreceptor model. Med. Chem. Commun. 2, 181-184.
- Löwer, M., Geppert, T., Schneider, P., Hoy, B. Wessler, S. & Schneider, G. (2011) Inhibitors of Helicobacter pylori protease HtrA found by 'virtual ligand' screening combat bacterial invasion of epithelia. PLoS ONE 6, e17986.
- Geppert, T., Hoy, B., Wessler, S. & Schneider, G. (2011) Context-based identification of protein-protein interfaces and 'hot-spot' residues. Chem. Biol. 18, 344-353.
- Zander, J., Hartenfeller, M., Hähnke, V., Proschak, E., Besier, S., Wichelhaus, T. A. & Schneider, G. (2010) Multistep virtual screening for rapid and efficient identification of non-nucleoside bacterial thymidine kinase inhibitors. Chem. Eur. J. 16, 9630-9637.
- Klenner, A., Hartenfeller, M., Schneider, P. & Schneider, G. (2010) 'Fuzziness' in pharmacophore-based virtual screening and de novo design. Drug Discov. Today Technol. 7, e237-e244.
- Reisen, F., Weisel, M., Kriegl, J. M. & Schneider, G. (2010) Self-organizing fuzzy graphs for structure-based comparison of protein pockets. J. Proteome Res. 9, 6498-6510.
- Weisel, M., Kriegl, J. M. & Schneider, G. (2010) Architectural repertoire of ligand binding pockets on protein surfaces. ChemBioChem 11, 556-563.
- Hiss, J. A., Hartenfeller, M. & Schneider, G. (2010) Concepts and applications of ‘natural computing’ techniques in de novo drug and peptide design. Curr. Pharm. Des. 16, 1656-1665.
- Schneider, G. (2010) Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 9, 273-276.
|
|

Editor
Gabriele Costantino
Univ. of Parma, IT
Editorial Committee
Erden Banoglu
Gazi Univ., TR
Lucija Peterlin Masic
Univ. of Ljubljana, SI
Leonardo Scapozza
Univ. of Geneve, CH
Wolfgang Sippl
Univ. Halle-Wittenberg, DE
Sarah Skerratt
Pfizer, Sandwich, UK

Executive Committee
Gerhard F. Ecker President
Roberto Pellicciari Past Pres.
Koen Augustyns Secretary
Rasmus P. Clausen Treasurer
Javier Fernandez Member
Mark Bunnage Member
Peter Matuys Member

|

