|
|
Young researcher
Stuart Conway
 Winner of the EFMC Prize for a Young Researcher in Academia Winner of the EFMC Prize for a Young Researcher in Academia
The interface of chemistry and biology, whether it be medicinal chemistry or chemical biology
My interest in biological and medicinal chemistry stems from undergraduate studies at the University of Warwick, where I read for a degree in Chemistry with Medicinal Chemistry (1994-1997). Summer placements at SmithKline Beecham (Worthing) and Unilever (Sharnbrook) were my first experience of a research environment and provided an early indication that this was a path I wished to pursue. During this time, a medicinal chemistry course, taught by visiting professor Roger Newton (Glaxo Wellcome), caught my imagination and I became interested in the idea of designing small molecules that have a certain effect on a biological system. This interest, coupled with the enjoyment of my final year project in the lab of Dr Andrew Clarke, led me to undertake a PhD. I moved to the University of Bristol to work in the lab of Prof. Jeff Watkins FRS and Prof. David Jane (1997-2000). Although my role was predominantly that of a synthetic chemist, the group was in the Department of Pharmacology and consequently research was carried out in an inherently multidisciplinary environment. My project was funded by Eli Lilly (Windlesham) and focused on the development of selective antagonists for the G-protein-coupled group III metabotropic glutamate receptors (mGluRs). From a scientific perspective, this work resulted in the most selective group III mGluR antagonists at the time.1 I enjoyed working in an environment where the compounds that I had synthesised were rapidly evaluated and iterative cycles of synthesis and design were possible. Following my PhD, I moved back to a chemistry department to work in the group of Prof. Andrew Holmes FRS at the University of Cambridge (2001-2003). My research now focused on intracellular, rather than extracellular, signalling molecules with the aim being to make derivatives of the second messenger d-myo-inositol 1,4,5-trisphosphate (InsP3) and the variously phosphorylated phosphatidylinositol phosphates (PIPs). This work was again, collaborative and multidisciplinary, working with the groups of Dr Martin Bootman and Drs Len Stephens and Phil Hawkins at the Babraham Institute. My time at Cambridge in the Holmes group reinforced my interest in not just making molecules, but employing synthetic organic chemistry as a powerful tool to study biology. In 2003 I began my independent research career as a Lecturer in Bioorganic Chemistry at the University of St Andrews. Since this time my research has focused on harnessing organic chemistry to develop probes for biological systems.
Chemistry illuminating biology – developing small molecule probes for biological systems
Although it is comparatively recently that a more collaborative approach between the pharmaceutical industry and academia has become common, I have long held the view that academia has a key role to play in therapeutic target identification and validation, and in the development of molecular tools to study biological systems. Most of the work conducted in my group is aimed at employing synthetic chemistry to enable biological experiments or solve biological problems. I have retained an interest in inositol-based signalling molecules with a key aim being the development of antagonists for the InsP3 receptors (InsP3Rs). Despite significant advances in InsP3-related biology, the pharmacology of these key receptors is relatively under-developed. We have systematically evaluated the effect of substituting the 4- and 5-position phosphate groups of InsP3 with phosphate bioisosteres.2,3 This approach led to successful development of some inositol-based antagonists of InsP3Rs,3 but there is still a lack of membrane permeant tool compounds for these fundamental receptors. Another project that I started early in my independent career is the development of light-activated (caged) molecular tools. Although unlikely ever to be a delivery tool in vivo, these compounds allow exquisite temporal and spatial control over the release of biologically active compounds in vitro. In particular our development of a wavelength-orthogonal system, in collaboration with Prof. Nigel Emptage, in which the release of the neurotransmitters GABA and glutamate was placed under the control of different colours of light, has many exciting applications.4 A collaboration with Prof. Ian Booth at the University of Aberdeen has led to an interest in the bacterial potassium efflux system, Kef. With Ian and Prof. Tarmo Roosild, we have been involved in elucidating the molecular mechanism of Kef gating by glutathione and its derivatives.5 This knowledge will allow us to design small molecules that are able to activate and inhibit the Kef system, a project that is on going within the group.
In 2008 my research group and I moved to the Department of Chemistry at the University of Oxford. The strong biological and medical science departments at Oxford have allowed me to develop a number of very exciting and productive collaborations. These projects include work on hypoxia-activated compounds with Dr Ester Hammond, work on Ca2+ signalling with Prof. Antony Galione and research on tumour imaging with Dr Ahmed Ahmed. Our interaction with the Structural Genomics Consortium (SGC, http://www.thesgc.org) was initiated by Dr Tom Heightman and Prof. Stefan Knapp and continues with Dr Paul Brennan. This collaboration has focused on the general area of epigenetics,6 and in particular on the development of small molecule probes for the acetyl-lysine-binding bromodomains.7,8 Epigenetics deals with heritable changes in gene expression or phenotype that are stable between cell divisions (and sometimes generations) but do not involve changes in the underlying DNA sequence of the organism.6 Epigenetic modifications include both methylation of DNA and post-translational modification (PTM) of proteins, including histones. Lysine acetylation is an important protein PTM, which is effected by histone acetyltransfereases (HATs), removed by histone deacetylases (HDACs) and is recognised or “read” by bromodomains. In collaboration with the SGC, we have developed a range of compounds that bind to bromodomains and inhibit their interaction with acetylated lysine (KAc).9,10 This project started with work towards assay development identifying N-methyl-2-pyrrolidone (NMP) as a small and ligand-efficient fragment that binds to the CREB binding protein (CREBBP) bromodomain.11 The ultimately led to the identification of the 3,5-dimethylisoxazole moiety as an effective KAc mimic and the development a range of bromodomain inhibitors based on this group. This work has mainly employed the structure-based design approach and, in addition to developing compounds that are potentially useful tools to study the bromodomains, we have learnt some fundamental information about the structure of the bromodomains, particularly about the role of some crystallographically-observed water molecules. We aim to employ this knowledge to further develop small molecules that are able to selectively prevent the interaction of a given bromodomain with its KAc-containing binding partner. By developing small molecule probes for these exciting targets we hope to further stimulate interest and research in this area.
The projects described above exemplify the approaches taken in the group and our core interests. I am excited about continuing to work on the chemistry-biology interface and I hope to further engage with the pharmaceutical sector in collaborative projects. I would like to thank all of my collaborators for making our interdisciplinary projects great fun to work on. I am deeply grateful to members of my research group, both past and present, who have successfully converted ideas that are formulated on a whiteboard in my office into physical reality in the lab.
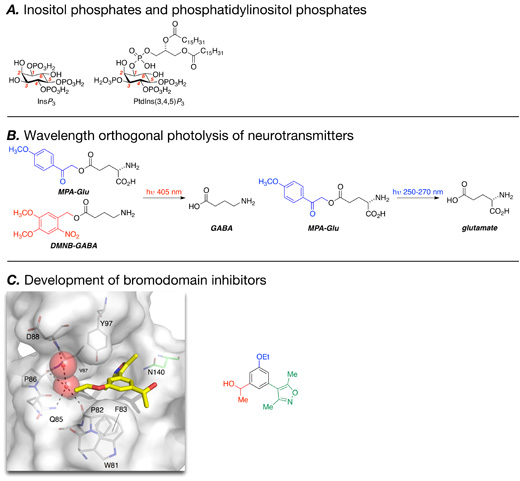
Figure 1. Examples of the molecules under investigation in the Conway group.
_________________________________________________________________
References
- Conway, S. J.; Miller, J. C.; Howson, P. A.; Clark, B. P.; Jane, D. E. Synthesis of Phenylglycine Derivatives as Potent and Selective Antagonists of Group III Metabotropic Glutamate Receptors. Bioorg. Med. Chem. Lett. 2001, 11, 777–780.
- Bello, D.; Aslam, T.; Bultynck, G.; Slawin, A. M. Z.; Roderick, H. L.; Bootman, M. D.; Conway, S. J. Synthesis and Biological Action of Novel 4-Position-Modified Derivatives of D-myo-Inositol 1,4,5-Trisphosphate. J. Org. Chem. 2007, 72, 5647–5659.
- Keddie, N. S.; Ye, Y.; Aslam, T.; Luyten, T.; Bello, D.; Garnham, C.; Bultynck, G.; Galione, A.; Conway, S. J. Development of Inositol-Based Antagonists for the D-myo-Inositol 1,4,5-Trisphosphate Receptor. Chem. Commun. 2011, 47, 242–244.
- Stanton-Humphreys, M. N.; Taylor, R. D. T.; McDougall, C.; Hart, M. L.; Brown, C. T. A.; Emptage, N. J.; Conway, S. J. Wavelength-Orthogonal Photolysis of Neurotransmitters in Vitro. Chem. Commun. 2012, 48, 657–659.
- Roosild, T. P.; Castronovo, S.; Healy, J.; Miller, S.; Pliotas, C.; Rasmussen, T.; Bartlett, W.; Conway, S. J.; Booth, I. R. Mechanism of Ligand-Gated Potassium Efflux in Bacterial Pathogens. Proc. Natl. Acad. Sci. USA. 2010, 107, 19784–19789.
- Arrowsmith, C. H.; Bountra, C.; Fish, P. V.; Lee, K.; Schapira, M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Disc. 2012, 11, 384–400.
- Hewings, D. S.; Rooney, T. P. C.; Jennings, L. E.; Hay, D. A.; Schofield, C. J.; Brennan, P. E.; Knapp, S.; Conway, S. J. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain-Acetyl-lysine Interactions. J. Med. Chem. 2012, 55, 9393–9413.
- Conway, S. J. Bromodomains: Are Readers Right for Epigenetic Therapy? ACS Med. Chem. Lett. 2012, 3, 691–694.
- Hewings, D. S.; Wang, M.; Philpott, M.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Vuppusetty, C.; Marsden, B.; Knapp, S.; Conway, S. J.; Heightman, T. D. 3,5-Dimethylisoxazoles Act As Acetyl-lysine-mimetic Bromodomain Ligands. J. Med. Chem. 2011, 54, 6761–6770.
- Brennan, P.; Hay, D.; Fedorov, O.; Filippakopoulos, P.; Martin, S.; Philpott, M.; Hewings, D.; Uttakar, S.; Heightman, T. D.; Conway, S.; Knapp, S. The design and synthesis of 5 and 6-isoxazolylbenzimidazoles as selective inhibitors of the BET bromodomains. Med. Chem. Commun. 2013, DOI: 10.1039/c2md20189e.
- Philpott, M.; Yang, J.; Tumber, T.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Keates, T.; Felletar, I.; Ciulli, A.; Knapp, S.; Heightman, T. D. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. BioSyst. 2011, 7, 2899–2908.
_________________________________________________________________
Sharan K. Bagal

Dr. Sharan K. Bagal (1978) graduated with first class honours in MChem in 2001 from the University of Oxford, U.K. where she subsequently completed her DPhil in chemistry in 2004 under the supervision of Professor Sir Jack Baldwin and Dr. Robert Adlington. Her doctorate was based on biomimetic natural product synthesis, in particular, the first total synthesis of two natural products biatractylolide and biepiasterolide via a biomimetic radical dimerisation. Sharan then moved to Professor Samir Zard’s group at the École Polytechnique in France as a postdoctoral researcher studying xanthate-based radical chemistry. This work led to the development of a novel acyl radical equivalent, along with a facile method for the generation of radicals from aldehydes.
In 2006 Sharan joined Pfizer Global Research and Development, Sandwich, UK as a medicinal chemist (www.pfizer.com), and is now a Senior Principal Medicinal Chemist at Pfizer Neusentis in Cambridge, UK (www.neusentis.com) with a focus on drug discovery targets for the treatment of pain. Sharan’s research interests encompass all aspects of medicinal chemistry and she has worked on several drug discovery targets including voltage-gated ion channels, GPCRs and protein kinases e.g. NaV1.8 and CXCR1/2. This work has used ligand-based medicinal chemistry design methods to identify potent and selective ion channel modulators within highly challenging medicinal chemistry space, and highly potent chemokine receptor antagonists with a balanced subtype selectivity profile. More recently, Sharan has used structure-based design methodology to successfully design highly potent receptor tyrosine kinase inhibitors with excellent selectivity and a favourable tissue distribution profile. Her focus has ranged from hit identification, hit to lead optimisation and candidate selection and Sharan has been directly involved in the invention of two compounds currently undergoing clinical trials.
So far over her career, Sharan has co-authored more than 20 papers and patents. She has a particular interest in aldehyde oxidase (AO) catalysed metabolism, in particular the structural aspects that underpin substrate recognition by AO and the role played by both steric and electronic effects of susceptible ring systems. She has developed several datasets that inform how structural features relate to species-specific metabolism by AO, and hypotheses of how this metabolism can be switched off by design. Sharan has also developed an interest in the design of compounds that penetrate, or are restricted from, the central nervous system (CNS) based on an understanding of CNS-located transporters and compound physicochemistry and the implications of CNS distribution on drug safety. She has delivered several reviews and presentations in this area.
|
|

Editor
Gabriele Costantino
Univ. of Parma, IT
Editorial Committee
Erden Banoglu
Gazi Univ., TR
Lucija Peterlin Masic
Univ. of Ljubljana, SLO
Leonardo Scapozza
Univ. of Geneve, CH
Wolfgang Sippl
Univ. Halle-Wittenberg, DE
Sarah Skerratt
Pfizer, Sandwich, UK

Executive Committee
Uli Stilz President
Gerhard F. Ecker Past Pres.
Koen Augustyns Secretary
Rasmus P. Clausen Treasurer
Hein Coolen Member
Gabriele Costantino Member
Phil Jones Member

|

